211service.com
Première simulation de niveau atomique d'une batterie entière
Lorsqu'il s'agit de développer la prochaine génération de technologies, le plus gros goulot d'étranglement est sans doute la batterie. Les ingénieurs ont besoin de meilleures batteries pour les véhicules électriques, pour le stockage d'énergie dans les réseaux électriques et, bien sûr, pour les appareils électroniques grand public.
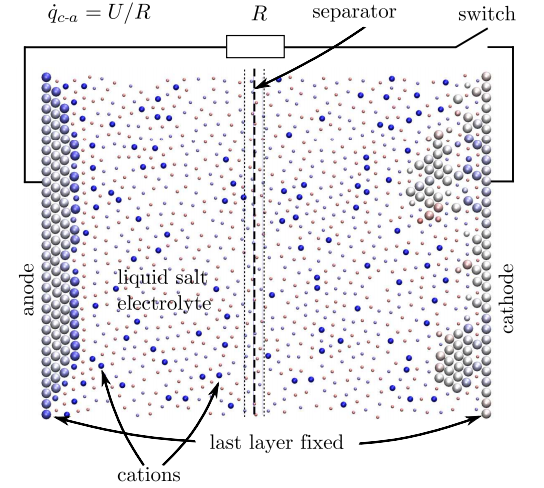
Ces batteries doivent fournir un courant plus élevé sur plus de cycles de décharge avec une plus grande densité d'énergie, pour ne citer que quelques-uns des défis.
Construire et tester de nouvelles conceptions de batteries prend du temps, est difficile et coûteux. Il est donc pratique pour les électrochimistes de simuler le fonctionnement d'une batterie avant même de se salir les mains.
C'est délicat. Personne n'a été capable de simuler une batterie entière au niveau atomique en raison de la complexité des processus en cours et des limites des techniques de modélisation actuelles.
Aujourd'hui, cela change grâce aux travaux de Wolf Dapp à l'Institute for Advanced Simulation et de Martin Muser à l'Université de la Sarre, tous deux en Allemagne. Ces gars-là ont simulé le comportement d'une batterie entière à l'échelle atomique. Et leur simulation reproduit pour la première fois bon nombre des caractéristiques réelles d'une batterie à partir des premiers principes.
Ces dernières années, les informaticiens ont fait des progrès significatifs dans la simulation de divers aspects du comportement des batteries. Ces modèles fonctionnent à mésoéchelle – plus petits que les électrodes mais plus grands que les molécules. Les simulations reposent sur des données expérimentales pour modéliser des éléments tels que la conductivité ionique et électronique, les coefficients de diffusion, les densités de courant, les potentiels électrochimiques, etc.
Ces modèles présentent un sérieux inconvénient : ils ont peu de pouvoir prédictif lorsqu'il s'agit de nouveaux matériaux ou de combinaisons de matériaux pour lesquels les données expérimentales ne sont pas disponibles. Pour prédire le comportement de nouveaux matériaux, les électrochimistes doivent modéliser les batteries à l'échelle des atomes et des molécules.
C'est difficile car les techniques utilisées par les informaticiens pour simuler le comportement des atomes et des molécules ne conviennent pas aux batteries. Ces simulations sont conçues pour des systèmes en équilibre ou proches de celui-ci. Ils fonctionnent en égalisant le potentiel chimique ou en minimisant l'énergie du système. Cependant, la différence de potentiel chimique entre deux électrodes est précisément ce qui entraîne le transport de charge dans une batterie, disent Dapp et Muser.
Ainsi, pour modéliser une batterie dans son ensemble, le modèle informatique doit prendre en compte tout changement d'énergie ou de potentiel chimique à chaque étape du calcul. C'est exactement ce que Dapp et Muser ont fait. Dans leur modèle, la charge est une variable qui peut être échangée entre les atomes et entre les liaisons à chaque étape du calcul.
Les simulations qui en résultent sont petites mais impressionnantes. Leur nanobatterie est constituée de 358 atomes, dont 118 constituent les électrodes. La cathode est initialement recouverte d'une couche de 20 atomes avec 39 atomes ionisés positivement dissous dans l'électrolyte.
Le calcul procède ensuite par étapes au cours desquelles les atomes peuvent se déplacer et échanger des charges au fur et à mesure que le système évolue. L'ensemble de la simulation se compose d'environ 10 millions de ces étapes.
Les résultats sont remarquables car ils reproduisent en fait les courbes de décharge génériques de vraies batteries macroscopiques. Par exemple, une température de fonctionnement inférieure réduit la capacité effective de la batterie simulée. Et surtout, la simulation reproduit la façon dont les batteries ordinaires s'usent. Lors de la recharge, les performances de la batterie se dégradent légèrement et la morphologie de la surface de l'électrode change pendant le fonctionnement de la batterie, selon Dapp et Muser.
Ces gars-là disent que le travail à ce stade n'est qu'un modèle de preuve de principe et qu'il existe diverses façons de l'améliorer à l'avenir. Par exemple, ils modélisent l'électrolyte à l'aide de particules qui ont une charge fixe et ne peuvent donc pas l'échanger.
Ce n'est pas ainsi que les électrolytes fonctionnent dans les vraies batteries et c'est donc un défaut important de la nouvelle méthode. Mais Dapp et Muser ont l'intention de corriger cela. Cette idéalisation sera abandonnée dans les travaux futurs, disent-ils.
Dans l'ensemble, cela semble être un travail important. Ce type de modèle pourrait considérablement améliorer la puissance prédictive des simulations de batterie et ainsi aider les électrochimistes à économiser du temps, de l'énergie et de l'argent avant de commencer des expériences détaillées.
Le résultat final devrait être de meilleures batteries, mais il y a un long chemin à parcourir avant cela.
Réf : arxiv.org/abs/1308.3424 : Réactions d'oxydoréduction avec des potentiels empiriques : simulations de décharge de batterie atomistique