211service.com
La théorie mathématique de la communication de Shannon appliquée au séquençage de l'ADN
L'un des grands héros méconnus de la science du XXe siècle est Claude Shannon, ingénieur des célèbres laboratoires Bell à son apogée au milieu du XXe siècle. La contribution la plus durable de Shannon à la science est la théorie de l'information, qui sous-tend toute communication numérique.
Dans un article célèbre datant de la fin des années 40, Shannon pose le problème fondamental de la communication : reproduire, à un point de l'espace, un message qui a été créé à un autre. Le message est d'abord codé d'une certaine manière, transmis puis décodé.
Shannon a montré qu'un message peut toujours être reproduit à un autre point de l'espace avec une précision arbitraire à condition que le bruit soit inférieur à un certain niveau de seuil. Il a ensuite calculé la quantité d'informations pouvant être envoyées de cette manière, une propriété connue sous le nom de capacité de ce canal d'information.
Les idées de Shannon ont été largement appliquées à toutes les formes de transmission d'informations avec beaucoup de succès. Une voie particulièrement intéressante a été l'application de la théorie de l'information à la biologie – l'idée que la vie elle-même est la transmission d'informations d'une génération à l'autre.
Ce type de pensée est en cours, révolutionnaire et encore à ses débuts. Il y a beaucoup à venir.
Aujourd'hui, nous nous penchons sur un corollaire intéressant dans le domaine de la transmission de l'information biologique. Abolfazl Motahari et des amis de l'Université de Californie à Berkeley utilisent l'approche de Shannon pour examiner à quelle vitesse les informations peuvent être extraites de l'ADN en utilisant le processus de séquençage au fusil de chasse.
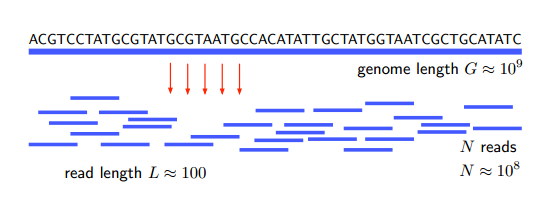
Le problème ici est de déterminer la séquence de nucléotides (A, G, C et T) dans un génome. Cela prend du temps car les génomes ont tendance à être longs – par exemple, le génome humain se compose d'environ 3 milliards de nucléotides ou de paires de bases. Cela prendrait une éternité à séquencer en série.
L'approche shotgun consiste donc à découper le génome en morceaux aléatoires, constitués de 100 à 1 000 paires de bases, et à les séquencer en parallèle. Les informations sont ensuite recollées in silico par un algorithme dit de réassemblage.
Bien sûr, il n'y a aucun moyen de savoir comment rassembler les informations à partir d'une seule lecture du génome. Ainsi, dans l'approche du fusil de chasse, ce processus est répété plusieurs fois. Étant donné que chaque lecture divise le génome d'une manière différente, les morceaux se chevauchent inévitablement avec les segments d'une analyse précédente. Ces zones de chevauchement permettent de réassembler l'ensemble du génome, à la manière d'un puzzle.
Cela ressemble à un problème classique de la théorie de l'information, et en effet diverses personnes ont pensé de cette manière. Cependant, Motahari et co vont plus loin en la reformulant plus ou moins exactement comme un analogue de la célèbre approche de Shannon.
Ils disent que le problème du séquençage du génome est essentiellement de reproduire un message écrit dans l'ADN, dans un format électronique numérique. Dans cette approche, le message original est dans l'ADN, il est codé pour la transmission par le processus de lecture, puis il est décodé par un algorithme de réassemblage pour produire une version électronique.
Ce qu'ils prouvent, c'est qu'il existe une capacité de canal qui définit un débit maximum de flux d'informations pendant le processus de séquençage. Il donne le nombre maximal de paires de bases d'ADN qui peuvent être résolues par lecture, par n'importe quel algorithme d'assemblage, sans tenir compte des limitations de calcul, disent-ils.
C'est un résultat important pour quiconque s'intéresse au séquençage des génomes. Une question importante est de savoir à quelle vitesse une technologie de séquençage particulière peut faire son travail et si elle est plus rapide ou plus lente que d'autres approches.
Cela n'est pas possible pour le moment car de nombreux algorithmes utilisés pour l'assemblage sont conçus pour des technologies et des approches de lecture spécifiques. Motohari et co disent qu'il existe au moins 20 algorithmes de réassemblage différents, par exemple. Cela rend difficile la comparaison de différents algorithmes, disent-ils.
Par conséquent, personne ne sait vraiment lequel est le plus rapide ou même qui a le potentiel d'être le plus rapide.
Le nouveau travail change cela. Pour la première fois, il devrait être possible de déterminer à quel point une technologie de séquençage donnée se rapproche de la limite théorique.
Cela pourrait bien forcer l'élimination du bois mort de cette zone et stimuler une période d'innovation rapide dans la technologie de séquençage.
Réf : arxiv.org/abs/1203.6233 : Théorie de l'information du séquençage de l'ADN