211service.com
Des biophysiciens découvrent quatre nouvelles règles de « grammaire » de l'ADN
Le biochimiste autrichien Erwin Chargaff est célèbre pour les deux règles qu'il a découvertes et qui portent désormais son nom. Au moment de cette découverte, en 1950, le plus gros problème en biologie était de comprendre la structure de l'ADN. Les règles de Chargaff se sont avérées être un indice important dans ce puzzle.
Les biologistes savaient depuis longtemps que l'ADN était constitué de quatre molécules : l'adénine, la guanine, la thymine et la cytosine. Ils ont supposé que ces molécules se produisaient en quantité égale et ont rejeté toutes les mesures qui suggéraient le contraire comme des erreurs expérimentales.
Chargaff a montré par une mesure minutieuse que cette hypothèse était fausse. Il a découvert que la quantité d'adénine était égale à celle de la thymine et que la quantité de guanine était égale à celle de la cytosine, mais qu'elles n'étaient pas égales l'une à l'autre. Les chiffres approximatifs sont : A=T=30 % et G=C=20 %.
La première règle de parité de Chargaff, comme on l'appelle maintenant, était un indice important que James Watson et Francis Crick ont utilisé pour développer leur modèle de paires de bases pour la structure en double hélice. Les biologistes savent maintenant que puisque A se lie à T et G se lie à C pour former une double hélice, cette règle s'applique à tout ADN double brin.
Chargaff a ensuite découvert qu'une version approximative de sa règle s'appliquait également à la plupart (mais pas à la totalité) de l'ADN simple brin. C'est beaucoup plus un casse-tête et les biologistes ne savent toujours pas pourquoi c'est vrai.
Les règles de Chargaff sont importantes car elles renvoient à une sorte de grammaire de la biologie, un ensemble de règles cachées qui régissent la structure de l'ADN. Cette grammaire devrait se révéler sous la forme de modèles dans l'ADN qui sont invariants pour toutes les espèces.
Mais au cours des 60 années écoulées depuis que Chargaff a découvert ses modèles invariants, aucun autre n'a émergé. Jusqu'à maintenant.
Aujourd'hui, Michel Yamagishi au Laboratoire de bioinformatique appliquée au Brésil et Roberto Herai à Unicamp à Sao Paulo, disent avoir découvert plusieurs nouveaux modèles qui élargissent considérablement la grammaire de l'ADN.
Leur approche est simple. Ces gars-là utilisent la théorie des ensembles pour montrer que les règles existantes de Chargaff impliquent l'existence d'autres modèles d'ordre supérieur.
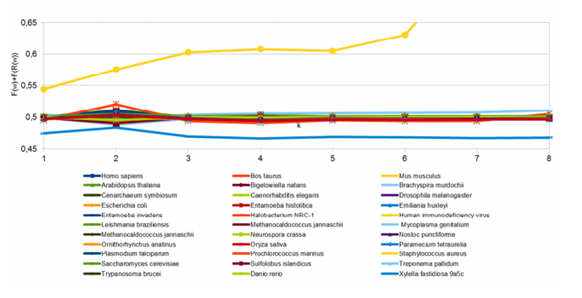
Voici comment. Une façon de penser aux modèles de l'ADN est de diviser une séquence d'ADN en mots de longueur spécifique, k. Les règles de Chargaff s'appliquent aux mots où k=1, en d'autres termes, aux nucléotides simples.
Mais qu'en est-il des mots avec k=2 (par exemple AA, AC, AG, AT et ainsi de suite) ou k=3 (AAA, AAG, AAC, AAT et ainsi de suite) ? Les biochimistes appellent ces mots des oligonucléotides. La théorie des ensembles implique que les ensembles entiers de ces k-mots doivent également obéir à certains modèles de type fractal.
Yamagishi et Herai les distillent en quatre équations.
Bien sûr, il n'est possible de voir ces modèles que dans d'énormes ensembles de données ADN. Effectivement, Yamagishi et Herai ont chiffré les séquences d'ADN de 32 espèces à la recherche de ces nouveaux modèles fractals. Et ils les ont trouvés.
Ils disent que les modèles apparaissent avec une grande précision chez 30 de ces espèces, y compris les humains, e coli et la plante arabidopsis. Seuls le virus de l'immunodéficience humaine (VIH) et Xylella fastidiosa 9a5c, un insecte qui attaque les pêches, ne sont pas conformes.
Ces nouvelles règles montrent pour la première fois que les fréquences des oligonucléotides ont des propriétés invariantes sur un large ensemble de génomes, disent-ils.
Cela pourrait s'avérer extrêmement utile pour évaluer les performances des nouvelles technologies de séquençage de génomes entiers à grande vitesse.
Un problème avec ces techniques est de savoir avec quelle précision elles fonctionnent. Yamagishi et Herai suggèrent qu'un test simple consisterait à vérifier si les génomes nouvellement séquencés contiennent ces modèles invariants. Si ce n'est pas le cas, c'est un signe que la technologie peut introduire une sorte de biais.
C'est un peu comme un test de somme de contrôle pour repérer les erreurs accidentelles dans les blocs de données et un bon morceau de science pour démarrer.
Réf : arxiv.org/abs/1112.1528 : Grammaire de la biologie de Chargaff : nouvelles règles de type fractal